Projects
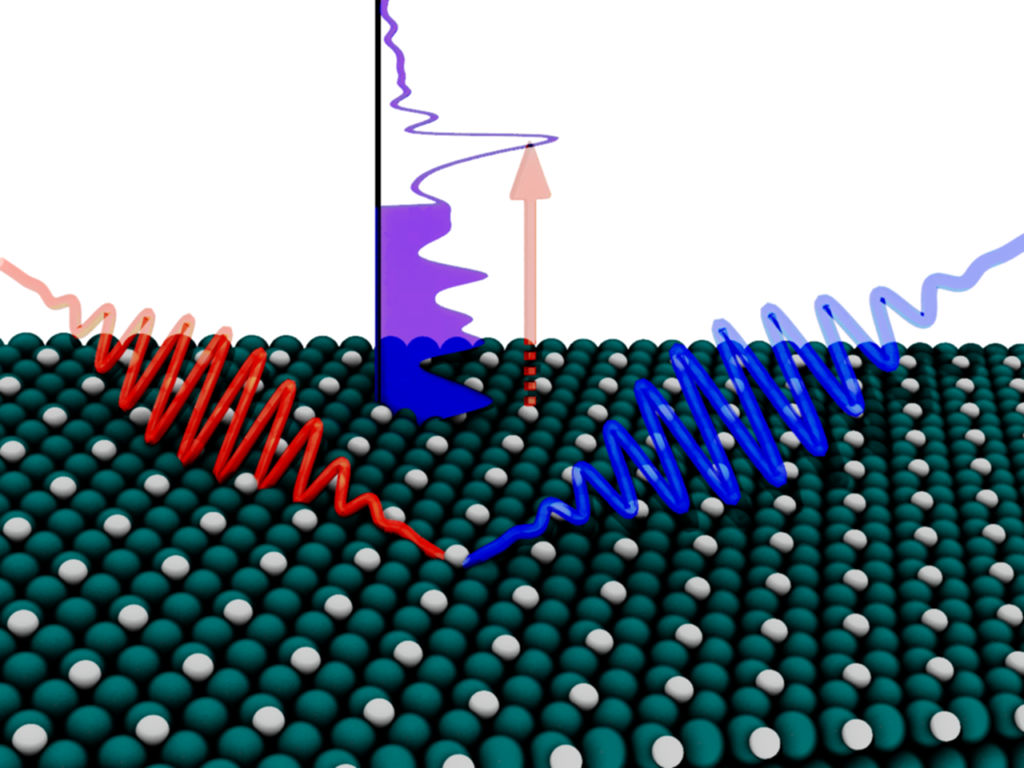
Ultrafast Catalysis
Simulating and understanding charge transfer and ultrashort-lived excitations in surface femtochemistry.
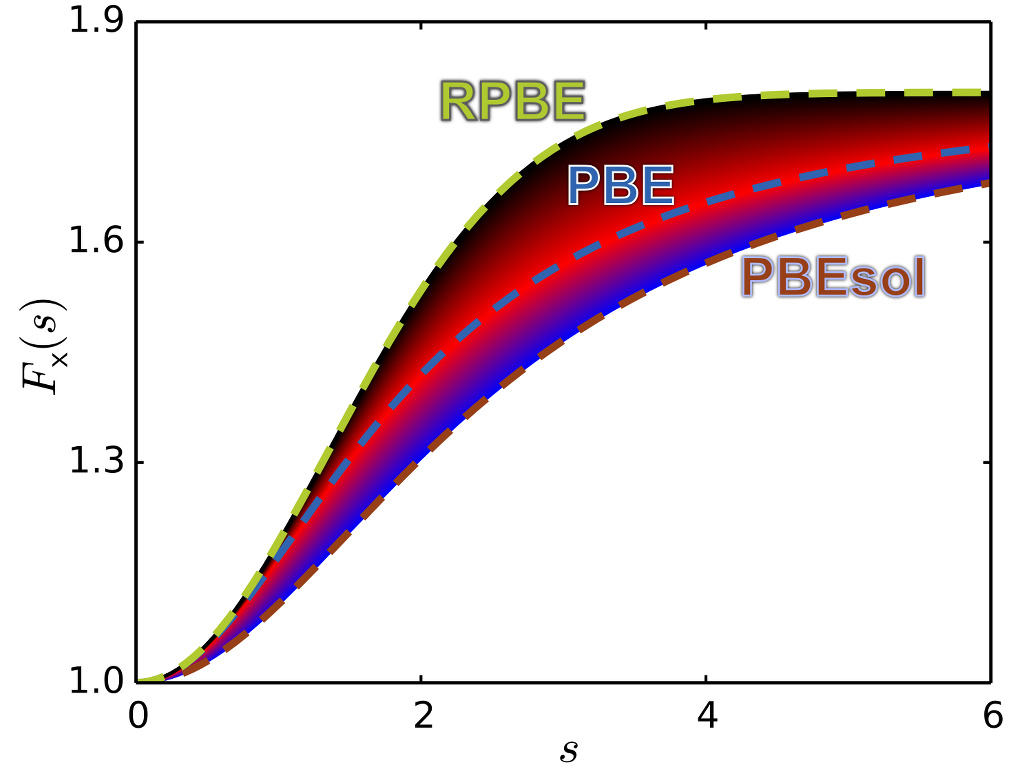
Exchange-correlation Functionals
Development of exchange-correlation functionals with improved description of bulk thermodynamics and surface reaction energetics.
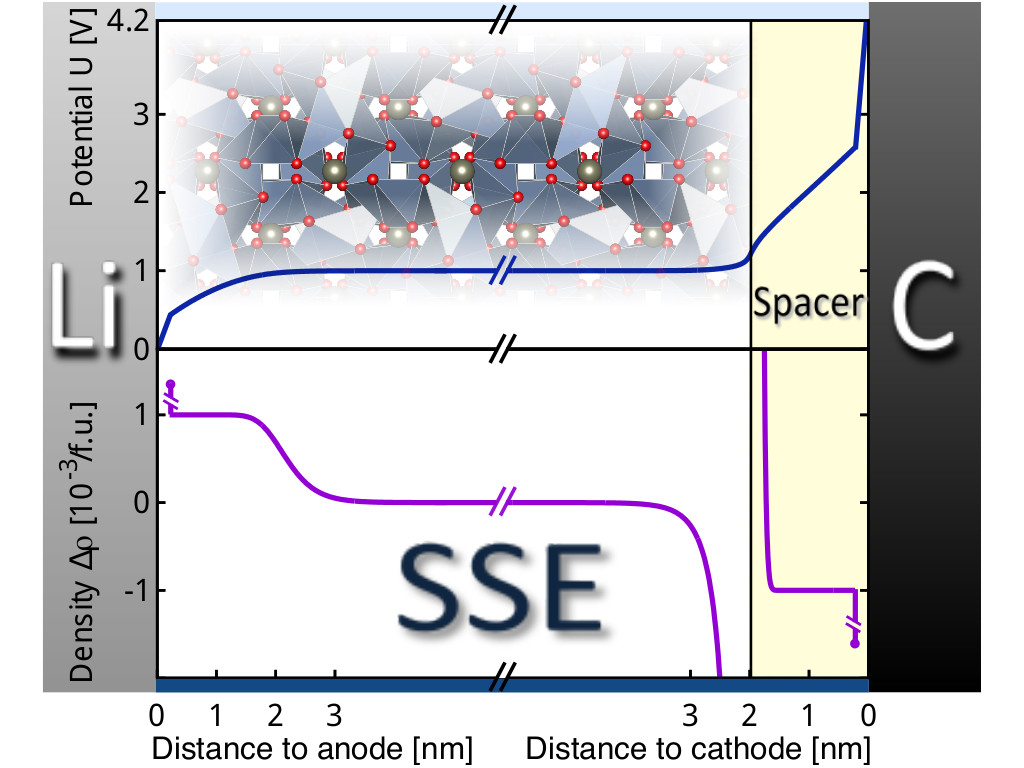
Solid-state Batteries
Modeling of solid-state electrolyte and inter-phase stabilities and charge double layers at electrode interfaces for potential all-solid-state Li-ion batteries.
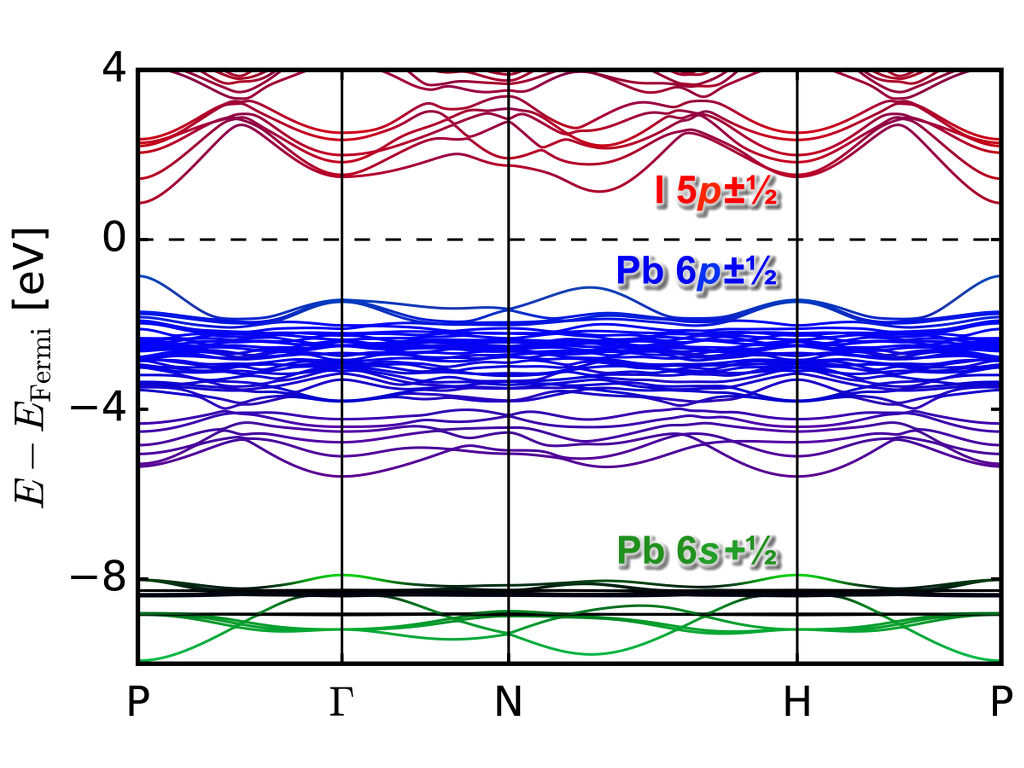
Perovskite Light Absorbers
Method development and computational search for new perovskite structure light absorbers for use in solar cells or as water-splitting photocatalysts.
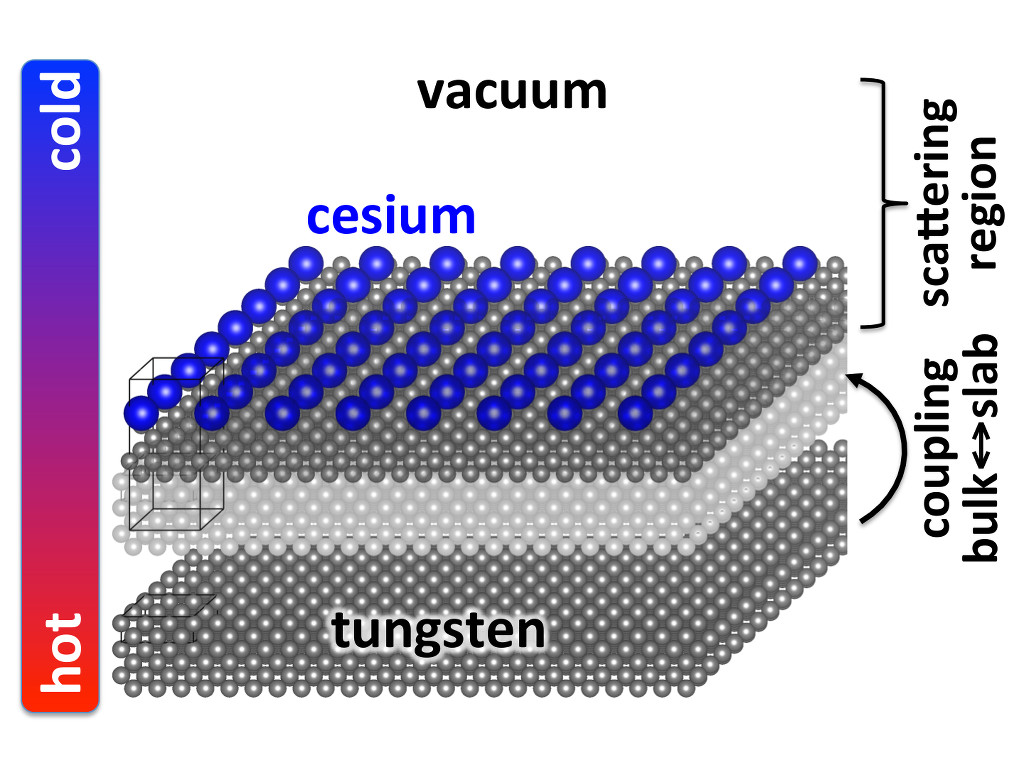
Thermionic Emission
Ab initio-based design of new stable materials with high thermionic emission currents for use as energy converters or cathodes.

Computational Lattice Dynamics
Simulation of ionic diffusion in energy storage materials and efficient calculation of phonon free energies for materials stability predictions.