
A yeast functional screen predicts new ALS disease genes
On this page:
Contents
Thank you for your interest. You will find on this webpage supplementary informations for this poster.
Abstract
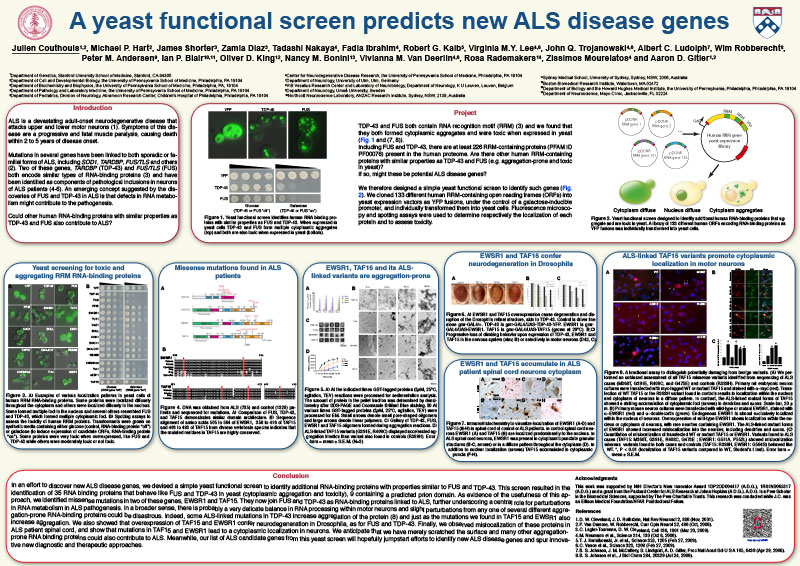
ALS is a devastating adult-onset neurodegenerative disease that attacks upper and lower motor neurons. Symptoms of this disease are a progressive and fatal muscle paralysis, causing death within 2 to 5 years of disease onset. Mutations in several genes have been linked to both sporadic or familial forms of ALS, including SOD1, TARDBP, FUS/TLS and others. Two of these genes, TARDBP (TDP-43) and FUS/TLS (FUS) both encode similar types of RNA-binding proteins and have been identified as components of pathological inclusions in neurons of ALS patients. An emerging concept suggested by the discoveries of FUS and TDP-43 in ALS is that defects in RNA metabolism might contribute to the pathogenesis. Could other human RNA-binding proteins with similar properties as TDP-43 and FUS also contribute to ALS?
Poster PDF
You can download a PDF copy of the "A yeast functional screen predicts new ALS disease genes" poster.
A yeast functional screen predicts new candidate ALS disease genes.
Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D, Epstein J, Chiang A, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Solski JA, Williams KL, Mojsilovic-Petrovic J, Ingre C, Boylan K, Graff-Radford NR, Dickson DW, Clay-Falcone D, Elman L, McCluskey L, Greene R, Kalb RG, Lee VM, Trojanowski JQ, Ludolph A, Robberecht W, Andersen PM, Nicholson GA, Blair IP, King OD, Bonini NM, Van Deerlin V, Rademakers R, Mourelatos Z, Gitler AD.
Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating and universally fatal neurodegenerative disease. Mutations in two related RNA-binding proteins, TDP-43 and FUS, that harbor prion-like domains, cause some forms of ALS. There are at least 213 human proteins harboring RNA recognition motifs, including FUS and TDP-43, raising the possibility that additional RNA-binding proteins might contribute to ALS pathogenesis. We performed a systematic survey of these proteins to find additional candidates similar to TDP-43 and FUS, followed by bioinformatics to predict prion-like domains in a subset of them. We sequenced one of these genes, TAF15, in patients with ALS and identified missense variants, which were absent in a large number of healthy controls. These disease-associated variants of TAF15 caused formation of cytoplasmic foci when expressed in primary cultures of spinal cord neurons. Very similar to TDP-43 and FUS, TAF15 aggregated in vitro and conferred neurodegeneration in Drosophila, with the ALS-linked variants having a more severe effect than wild type. Immunohistochemistry of postmortem spinal cord tissue revealed mislocalization of TAF15 in motor neurons of patients with ALS. We propose that aggregation-prone RNA-binding proteins might contribute very broadly to ALS pathogenesis and the genes identified in our yeast functional screen, coupled with prion-like domain prediction analysis, now provide a powerful resource to facilitate ALS disease gene discovery.
References
PNAS December 27, 2011 vol. 108 no. 52 20881-20890
Pubmed: PMID 22065782 DOI: 10.1073/pnas.1109434108
Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis.
Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, Kim CE, Frackelton EC, Solski JA, Williams KL, Clay-Falcone D, Elman L, McCluskey L, Greene R, Hakonarson H, Kalb RG, Lee VM, Trojanowski JQ, Nicholson GA, Blair IP, Bonini NM, Van Deerlin VM, Mourelatos Z, Shorter J, Gitler AD.
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease affecting motor neurons. Mutations in related RNA-binding proteins TDP-43, FUS/TLS and TAF15 have been connected to ALS. These three proteins share several features, including the presence of a bioinformatics-predicted prion domain, aggregation-prone nature in vitro and in vivo and toxic effects when expressed in multiple model systems. Given these commonalities, we hypothesized that a related protein, EWSR1 (Ewing sarcoma breakpoint region 1), might also exhibit similar properties and therefore could contribute to disease. Here, we report an analysis of EWSR1 in multiple functional assays, including mutational screening in ALS patients and controls. We identified three missense variants in EWSR1 in ALS patients, which were absent in a large number of healthy control individuals. We show that disease-specific variants affect EWSR1 localization in motor neurons. We also provide multiple independent lines of in vitro and in vivo evidence that EWSR1 has similar properties as TDP-43, FUS and TAF15, including aggregation-prone behavior in vitro and ability to confer neurodegeneration in Drosophila. Postmortem analysis of sporadic ALS cases also revealed cytoplasmic mislocalization of EWSR1. Together, our studies highlight a potential role for EWSR1 in ALS, provide a collection of functional assays to be used to assess roles of additional RNA-binding proteins in disease and support an emerging concept that a class of aggregation-prone RNA-binding proteins might contribute broadly to ALS and related neurodegenerative diseases.
References
Hum. Mol. Genet. (2012) 21 (13): 2899-2911
Pubmed: PMID 22454397 DOI: 10.1093/hmg/dds116
Contact
Have a look on my contact informations page.
Last modified Thuesday, 9-Sep-2014 15:18:32 PDT