

|
 |
 |

|
 |
|

|

|
 |
|
 |
Clinical Features
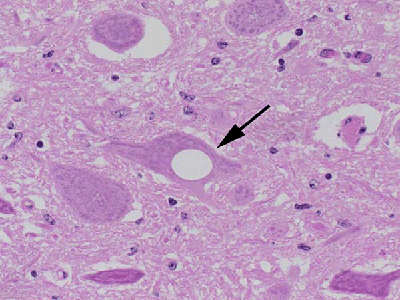 | Examination of brain tissue reveals florid plaques of agglomerated prions and vacuolization. |
| http://pathology.mc.duke.edu/neuropath/CNSlecture2/cjd.jpg |
| Incubation | Long incubation periods of up to 30 years |
| Outcome | Symptoms are progressive and result in fatality |
| Incubation | Long incubation periods of up to 30 years |
| Pathology | Prions cause spongiform degeneration, a process of vacuolization in which the brain deteriorates and looks spongy |
| Symptoms | Behavorial effects include ataxia, peripheral sensory changes and dementia |
| Diagnosis | Diseases are invariably fatal, and definite diagnosis can usually only be made at autopsy |
| Human Prion Diseases | Animal Prion Diseases |
| Creutzfeldt-Jakob Disease (CJD) Variant Creutzfeldt-Jakob Disease (vCJD) Gerstmann-Straussler-Scheinker Syndrome Fatal Familial Insomnia Kuru |
Bovine Spongiform Encephalopathy (BSE) Scrapie Chronic Wasting Disease (CWD) Transmissible mink encephalopathy Feline spongiform encephalopathy Ungulate spongiform encephalopathy |
Human TSEs occur in sporadic, familial, and acquired forms. The most common form, sporadic Creutzfeldt-Jakob disease (CJD), has a worldwide death rate of about 1 case per million people each year and is responsible for about 85% of all CJD cases worldwide (WHO). Familial Creutzfeldt-Jakob disease (fCJD) is inherited as a result of genetic mutations and counts for 10-15% of the cases worldwide (WHO). The remaining cases are iatrogenic (accidental transmission through contaminated surgical equipment, as a result of cornea or dura mater transplants, or due to the administration of human growth hormones) and variant Creutzfeldt-Jakob disease (WHO).