At the very basis of the computational design of materials lies the prediction of the stability: can a material with predicted desired (optical, electronic, catalytic, etc.) properties be at all synthesized in the lab or could it even withstand working conditions?
In this project, the thermodynamic stability, phase transitions, and decomposition reactions of crystal structures are studied. The elevated temperature behavior of (ideal) structures is modeled on the basis of the phonon gas. The computational effort for the calculation of the phonon modes within density functional or density functional perturbation theory scales with the fourth power of the system size: these calculations are thus more demanding than T=0K ground state calculations.
Fast Phonon Free Energy Estimates
If relatively rough estimates of phonon free energies (e.g. for screening studies) are sufficient, an approximate method developed here can be used yielding phonon frequencies at basically the same cost as a DFT ground state calculation. The phonon calculations are accelerated by first calculating the force constant matrix using a simply model Hamiltonian allowing for computations with negligible effort compared to DFT [Voss and Vegge, J. Chem. Phys. 128, 184708 (2008)].Any kind of force field method could be used to provide the model Hamiltonian. We use here a Bader decomposition of the electronic charge density and use Ewald summation to calculate forces for the corresponding point charge Hamiltonian.
The eigenvectors $|i\rangle$ of the model force constant matrix are summed up with arbitrary phases:
$$ |w\rangle = \sum_{i=1}^{3N} |i\rangle .$$
With a single DFT calculation, the force response to $|w\rangle$ as a displacement (with reasonably scaled length) from the equilibrium coordinates is obtained. Assuming that the $|i\rangle$ were eigenvectors of the correct DFT dynamical matrix, the eigenvalues $k_i$ of the Hessian $H$ can be reconstructed (note that $H |w\rangle$ is estimated using only a single DFT calculation)
$$ k_i = \langle i | \big( H | w \rangle \big) .$$
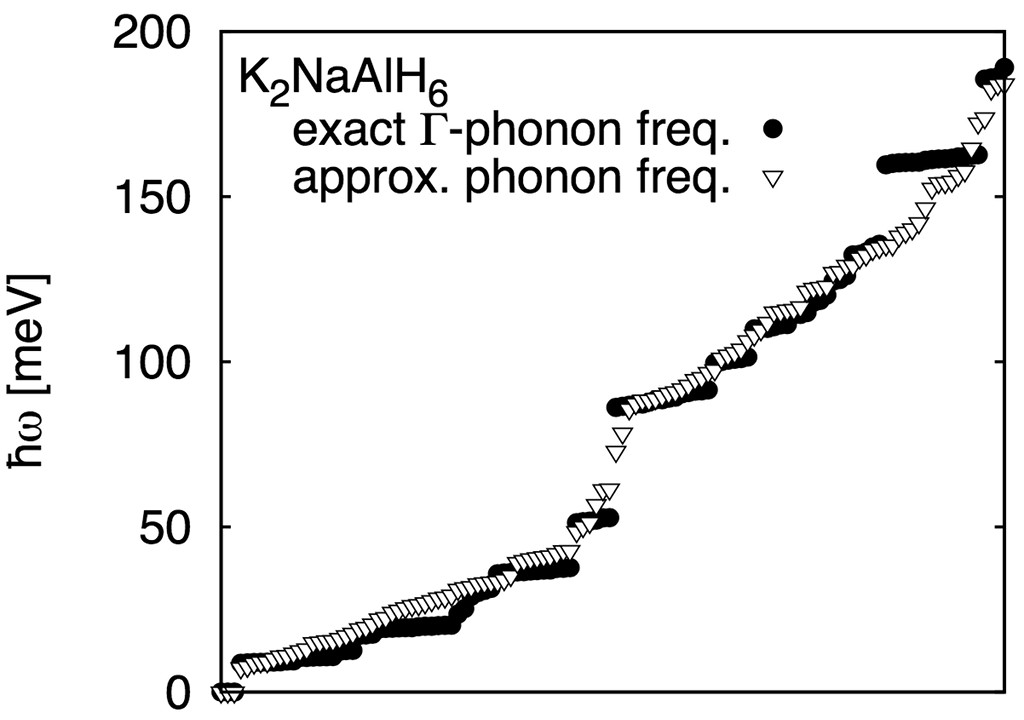
From the approximate eigenpairs $k_i,|i\rangle$, the Hessian can be reconstructed in the canonical basis of the atomic coordinates, and the dynamical matrix can be calculated via mass-scaling. The resulting phonon spectrum is only approximate; degeneracies, e.g., of the accurate spectrum are generally not reproduced:
The phonon free energy is given by an integral over the phonon density of states $g(\omega)$
$$ F_{\rm phon} = 3N\,kT \int\limits_0^\infty {\rm d} \omega \, g(\omega) \,{\rm ln}\left[2 {\rm sinh}\left(\frac{\hbar \omega}{2kT}\right)\right] ,$$
and predicted reaction temperatures are thus in relative agreement with the more expensive DFT-phonon calculation results:
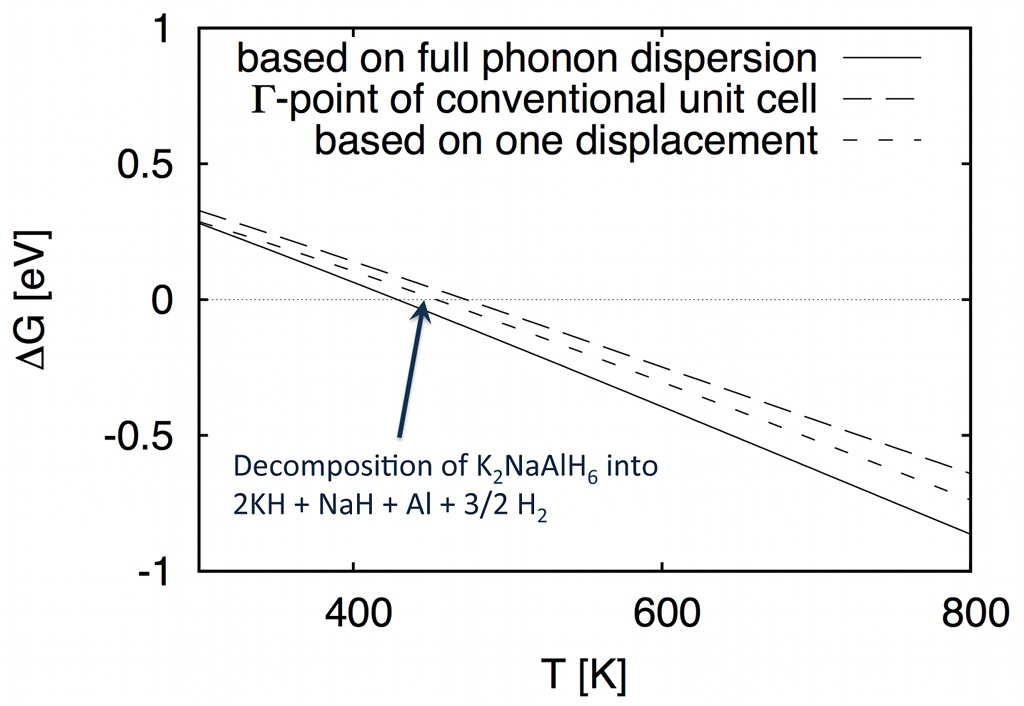
This fast phonon method thus allows for efficient screening of the thermodynamic stability for a large phase space of materials combinations to select materials of potential interest that can then be studied in more detail and with higher accuracy.
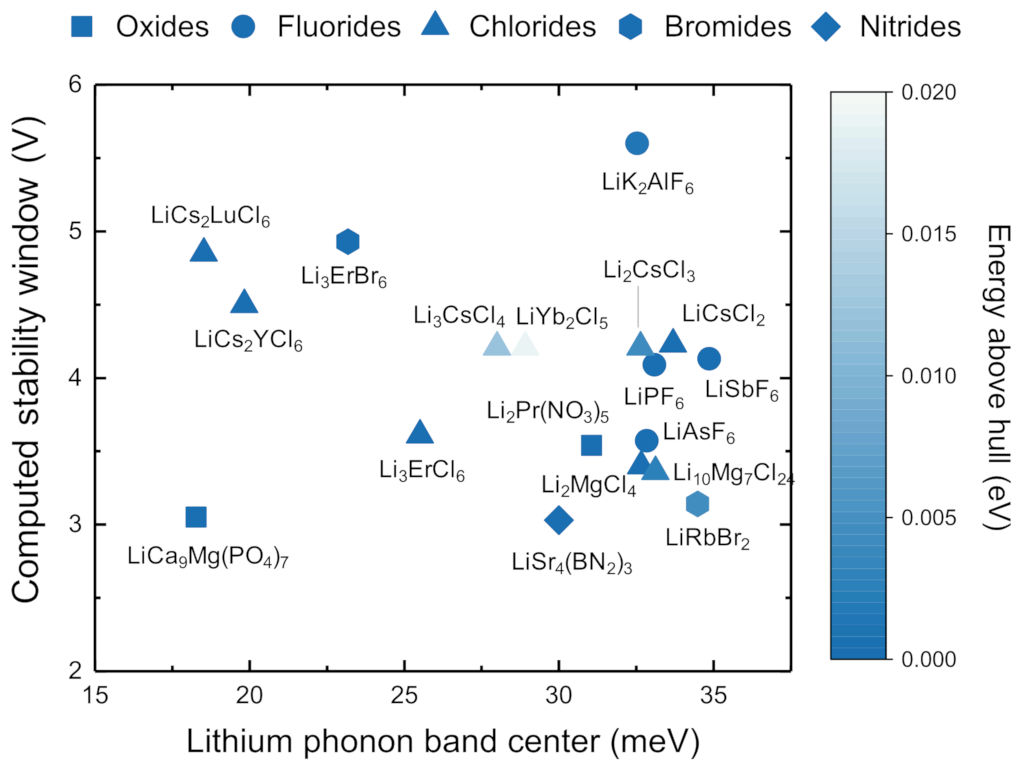
Correlating average phonon energies with ionic mobility, the above method has been used to screen 14,000 Li-containing compounds for their potential use as super-ionic conductors in solid state batteries [Muy, Voss, Schlem, Koerver, Sedlmaier, Maglia, Lamp, Zeier, Shao-Horn, ISCIENCE 16, 270 (2019)].
Displacive Phase Transitions
For the prediction of phase transitions due to phonon frequencies becoming imaginary (i.e. there is no force driving the system back to equilibrium for these modes), more accurate phonon calculations than what the above method can provide are needed. These calculations can be based on density functional perturbation theory or frozen phonon calculations (both involving $O(N)$ times the effort of a single DFT calculation).
The figure above shows the density functional theory results for phase stabilities of Mg(BH4)2. A highly symmetric prototype structure of $I \bar 4m_2$ symmetry is found to be susceptible to a distortion due to a lattice strain instability (irreducible representation $\Gamma_4$) to the $F222$ phase, which turns out to be more stable at elevated temperatures, too [Voss et al., J. Phys.: Condens. Matter 21, 012203 (2008)].
Solid State Rate Processes
Explicitly simulating thermally activated processes by means of first principles molecular dynamics calculations typically involves computationally prohibitively long timescales required for these often rare events to occur. Instead, the trajectory of most statistical weight, the so-called minimum energy path, can be sampled using a finite number of replica or images of the system.
We use the nudged elastic band method [Jónsson, Mills, and Jacobsen, in Classical and Quantum Dynamics in Condensed Phase Simulations, ed. by Berne, Ciccotti, and Coker, pp. 385-404 (World Scientific, Singapore, 1998)] to find minimum energy paths based on initial guesses for the reaction paths. Shown below are two such optimized paths for vacancy-mediated proton diffusion in Na3AlH6.
The density functional theory results for energy barriers for diffusion through the crystal by hopping between adjacent AlH6 octahedra has a much higher barrier than vacancy jumps within the same octahedron [Voss et al., J. Phys. Chem. B 111, 3886 (2007)], which is in good agreement with quasi-elastic neutron scattering studies [Shi, Voss, et al., J. Alloys Compd. 446, 469 (2007)]. The higher barriers for long-range type diffusion unfortunately limit the capability of this material to release or absorb hydrogen, such that sodium alanate does not have practical use as a lightweight hydrogen storage material.