Research in this area focusses on the development of a multi-scale model describing the electrochemistry in proton exchange membrane fuel cells (PEMFCs). Chemical and electrochemical reactions in PEMFCs occur on nano-size platinum particles. The multi-scale model is based on dynamic Monte Carlo (DMC) simulations of the chemistry on the surface of these particles. This allows considerations of specific chemical effects such as the influence of adatom interactions and active sites. The transition probabilities required for the DMC simulations are taken from literature data or determined from quantum simulations. To ultimately validate the simulations, experiments are performed under well-controlled conditions.
PEMFCs are touted to be the energy systems in the future for a wide range of applications including automobiles and portable devices. Performance of PEMFCs need to be improved significantly to make them economically competitive with conventional energy systems, like IC engines. Slow electrochemical reactions at the cathode cause huge potential losses in PEMFCs. The purpose of this research is to unravel the cause of these losses by understanding the underlying phenomena at atomic scales. We are using a combination of Monte Carlo and ab-initio simulations, and micro-scale experiments for this purpose.
Research areas are divided into quantum simulations, dynamic Monte Carlo simulations, and experimental analysis.
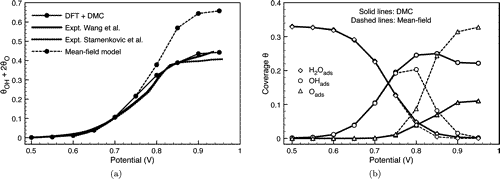
First-Principles Analysis of Oxygen-Containing Adsorbates Formed from the Electrochemical Discharge of Water on Pt(111)
(Published in J. Phys. Chem. C, 112 (26), 9760-9768 2008)
Adsorbent Interactions in Electron-Transfer Reactions Electron-transfer:
Electron-transfer with OH interaction:
OHads-OHads + H+aq + e- → OHads-OH2,ads
Hydrogen-hopping due to OH interaction:
OHads-OH2,ads → OH2,ads-OHads
(Presented in 209th Electrochemical Society Meeting)

Convergent Iterative Constrained Variation Method to Locate Transition States of Electron-Transfer Reactions
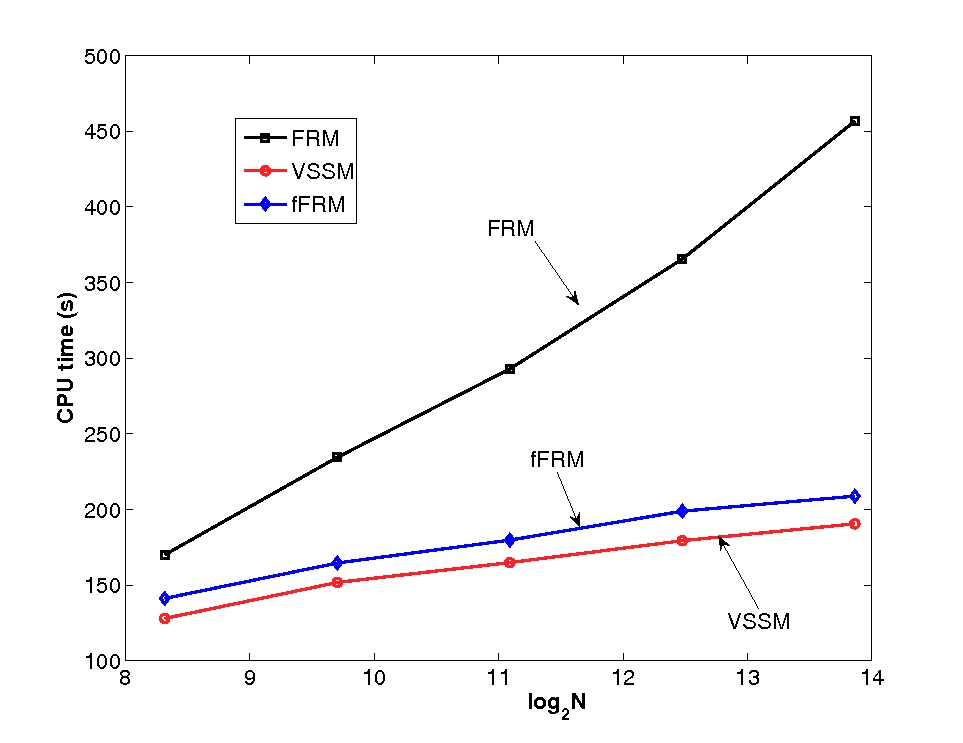
Efficient dynamic Monte Carlo algorithm for time-dependent catalytic surface chemistry
(Published in Phys Rev E. 2006 74, 046707)

Dynamic Monte Carlo (DMC) Simulations
DMC is a numerical method to address very high-dimensional complex physical problems using random sampling, with applications in interfacial and non-equilibrium chemistry, like that in automobile catalytic converters. It is a stochastic model of the system, which simplifies the description of the system, as well as a numerical solution of the problem of relative ease. DMC has uses for PEMFCs because PEM fuel cell electrochemistry is inherently stochastic in nature; an accurate treatment using DMC is possible and therefore a natural choice, although it does have its challenges. Platinum catalyst nanoparticles have a cubo-octahedral 3D geometry with eight (111) and six (100) faces. Moreover, catalytic properties vary significantly from the atoms located on the faces to those located on the edge or corners of the nanoparticle. So far, a DMC method has never been applied to such complex geometries due to very complicated implementation. Results so far show that DMC simulations can be extended to 3D geometries, and preliminary results demonstrate capability to capture important kinetic features.
(Provided by Varun Rai)


Features:
- Pt single crystal ultra-microelectrode fabrication
- Determination of kinetic rate parameters for O2 reduction reaction on preferentially oriented Pt surface
- Analysis of scaling of the triple-phase boundary (TPB) region
- Validation of DMC simulation
This research combines both computational and experimental tools to study the limitations of PEM fuel cells due to slow electrochemical reactions. The kinetic rate data are computed by quantum calculations, which are used as input to the DMC simulations. The experiments serve as validation for the DMC simulations, besides providing fundamental understanding of the electrochemical phenomena at the electrodes of a PEM fuel cell. DMC simulations for non-equilibrium surface chemistry were extended to 3D nanoparticle geometries. Capability to capture important phenomena in such complex electrochemical system was also demonstrated. Quantum simulations were used to study the mechanisms for different reactions on catalyst surfaces and for computing kinetic rate data. Preliminary investigations of O2 reduction kinetics at Pt/Nafion interface were performed using standard electrochemical techniques. A new approach for the fabrication of Pt single crystal ultra-microelectrodes using FIB is suggested.
(Provided by Abhishek Dhanda)