 |
 |
Peptide Library Mini-Tutorial:
For several years we have been developing the tools to using cells as individual test tubes for the creation and analysis of small peptides (generically termed here DOMINANT EFFECTORS or DEs) that act upon signaling pathways of choice.
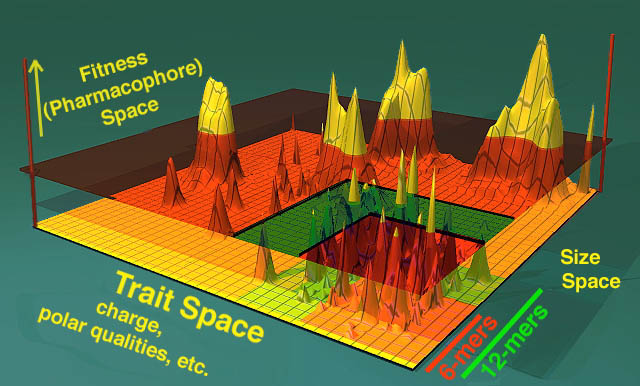
Understanding how peptide libraries work is rooted on the premise of shape-space, an artificial mathematical place where you can consider the universe of all potential peptides to be represented on one axis of a 3-dimensional world (see below, y axis with 6-mers and 12-mers). In a second axis one can imagine the peptides so encoded have varying charges, shapes, and chemical features. In yet another axis (in this example the Z axis) one represents the fitness of the peptide to carry out a given task (ie bind to some intracellular target protein and change that protein's function).
The X-Y plane of features as shown below represent a universe of shapes (a subset of which includes peptides in the lower left hand corner) and their fitness at a given task is represented in the Z axis. The peaks represent "clouds" of molecules that are similar to each other in sequence and size and are, as well, capable of carrying out the defined task. The better they are at the task, the higher they fall on the Z axis. This is termed a fitness landscape.
The transparent orange glass that sits parallel to the X-Y plane represents the "selection cutoff". All peptides above the selection cutoff plane are hits and all peptides below the plane are undesired events. Fitness is also named "pharmacophore space" as it represents the subset of shapes that can have pharmacologic effects. Realize that each of these axes are artificial constructs and that shape space is as varied as the imagination can make it. For instance, one can imagine a parallel universe of overlapping organic molecules that have the exact same function as the peptides selected. The routes that lead from the shape space depicted and the universe of pharmacologically active, organic small-molecule drugs is currently unmapped, but the secret formulas DO exist... (muhahhahahaha!!).
Enough of that mathematical gibberish I wanna know about peptide libraries!
Specifically, screening libraries of peptides by their expression inside mammalian cells offers an opportunity to simultaneously interrogate intracellular signaling pathways, create reagents to further an understanding of the pathway, and to create novel forms of therapies. For us, it is a marriage of pharmacology with biochemistry and genetics in a single system.
By delivering libraries of peptides to cells we can scan for rare peptides that bind to and induce function of intracellular signaling proteins and surface molecules. Using these peptides as affinity reagents we can isolate their target protein complexes and discern mechanism. Finally, we are using them to create therapeutic modalities from either their targets, the peptides themselves, or by some better understanding of the biological process that is uncovered.
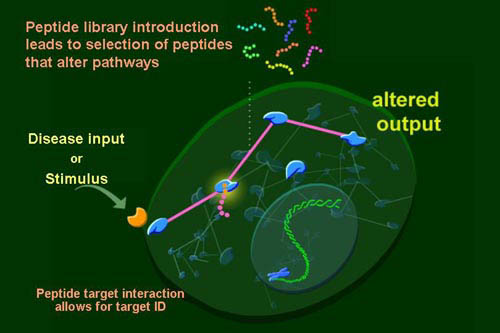
For the purposes of this web page tutorial on screening with peptides we need to be able to accomplish the following GENERIC outlined points in bold. The explanation after each bolded generic example gives a specific reference from our published paper on the approach. Other publications demonstrating the robustness of the approach are forthcoming.
1. Define a genetic screen that can distinguish cells expressing peptides with a desired phenotype from a background of cells not expressing a peptide of interest. For example, in our most extensive tests of searching within libraries for peptide inhibition of intracellular signaling we had undertaken a screen for investigation of taxol-induced cell death. Very little is understood about taxol-mediated cell death but it is known that taxol binds to tubulin, interferes with tubulin disassembly and arrest cells at G2/M. We set up selection conditions (Nature Genetics, Xu et al) in which taxol would induce significant death of HeLa cells, a cervical carcinoma in which p53 is inactivated through association with HPV E6 protein. 3 x 108 cells were infected with a library of constrained peptides (containing an 18 amino acid insert). A selection was set up to isolate clones that inhibit taxol-induced death.
2. Devise approaches to rescue the peptide inserts, transfer them to virgin cells and confirm the phenotype. Clones were isolated and PCR cloning of the peptide inserts was undertaken. The insert were recloned into tetracycline inducible constructs for further testing. 4 peptides were found to reproducibly protect cells from taxol mediated death over a range of taxol concentrations and with varying degrees of efficiency.
3. Be able to demonstrate the peptide binds a target structure (best if a protein) in mammalian cells that correlates with or explains its function. Clone the protein if necessary. Initial results with yeast-two hybrid using one peptide (h1a8-5.1) as bait suggests that this peptide binds to a proteosome subunit, (15 million yeast two hybrid clones were searched and this clone was found 7 times in the search out of 8 positive clones). A second peptide was screened and in yeast 2-hybrid a Bcl-X binding protein was identified as a target 15 out of 16 times (unpublished).
4. Demonstrate with orthoganal approaches that action upon processes controlled by this cloned/identified target by other approaches reproduces the phenotype conferred by the peptide or otherwise confirms its identification. The h1a8-5.1 peptide was tested the furthest to date. We further characterized the mechanism by which h1a8-5.1 blocks apoptosis. One general mechanism of resistance to taxol is genetic upregulation of MDR/ABCB1 protein pump activity. This peptide blocked apoptotic death and upregulated the MDR protein expression at the cell surfaceæ (numerous control peptides were used which showed no such effects). Pump activity is also upregulated as tested through loading of cells with rhodamine red and testing for pump efflux of this dye (a standard test for MDR activity) and by directly testing the expression of MDR protein at the cell surface.
5. Demonstrate that drug-like action upon the processes targeted recapitulate the phenotype. This peptide could regulate taxol-mediated death through a broader mechanism of gene activation of MDR pump activity or some other post-translational upregulation of pump activity. We suspected that perhaps the peptide might be inhibiting degradation of MDR protein. Tests with the a proteasome inhibitor MG132 show similar upregulation of the MDR protein but ALSO enhanced transcription of the MDR locus over peptide alone. Thus, we suspect that this peptide is acting through inhibition of a proteosome function possibly by blocking degradation of a repressor of MDR transcription. Further studies are needed in this area.
Scaffolds:
A variety of peptide presentation techniques can be considered. We have used both linear and constrained peptide scaffolds and both have given rise to peptides with functions. See the manuscript by Xu et al and Gururaja TL, et al (A novel artificial loop scaffold for the noncovalent constraint of peptides. Chem Biol. 2000 Jul;7(7):515-27.) for more information on scaffolded peptides. The following figure shows the linear and constrained formats we currently employ and that have given rise to successful screens. The pros and cons are shown on the right hand side of the figure.

In the creation of a library, a retroviral construct of the following general design is made:
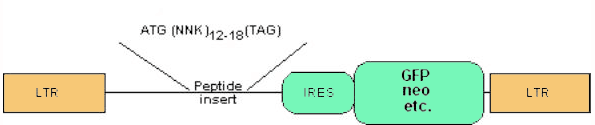
And the insert is formulated as follows: The peptide insert structures for the constructs is shown. Certain of these vectors are modified to include selectable markers or inducible promoters. Retrovirus vector with internal IRES sequence. Multiple different selectable markers have been placed into our constructs, including neomycin, blastocidin, bleomycin, puromycin resistance, and GFP genes as well as surface markers such as CD8.
-
It is important to understand that in any library system encoded by DNA oligonucleotide synthesis one cannot easily have complete control over the codons that will eventually be incorporated into the peptide structure (there are ways to completely control codon usage but they are beyond the scope of this tutorial). This is especially true in the case of codons encoding stop signals (TAA, TGA, TAG).
-
In a synthesis with NNN as the random region, there is a 3/64, or 4.69%, chance that the codon will be a stop codon.
-
Thus, in a peptide of 10 residues, there is an unacceptably high likelihood that 46.7% of the peptides will prematurely terminate. But for larger structures, such as those envisioned here, such termination will lead to sterile peptide expression.
-
To alleviate this, we encode random residues as NNK, where K= T or G.æ This allows for encoding of all potential amino acids (changing their relative representation slightly), but importantly preventing the encoding of two stop residues TAA and TGA.
-
Thus, libraries encoding a 10 amino acid peptide will have a 15.6% chance to terminate prematurely. NNK is a more acceptable alternative. For more details on construction of peptide libraries consult the article by Xu et al.
![]()
Related links:
Also of interest is the Nature Genetics manuscript on which the peptide library work was based.
Home Page |
Interests | Members
|
MTA Forms | Plasmid
Maps | Retroviral Systems
| Genetic Screens | Library
Systems | Protocols
| Tutorials |
Publications | Contact
| Virus Chat