-
Introduction
The earth's organisms are a vast repository of genetic diversity. Each
species (n >106) is distinguished from every other by a unique
genomic sequence that is passed on to successive generations with extremely
high, but not perfect, fidelity. Imperfections in DNA replication and repair
mean that the genome of each member of a species is also unique. Intraspecific
differences are one basis for individuality, including individual differences
in susceptibility to disease. The most striking example of such differences
are genetic diseases.
1.1 The Need For Animal Models And A New Approach
To Obtaining Them.
Animal models of genetic diseases have been extremely useful. Models
can arise from chance discoveries, such as narcopleptic dogs , or by intentional
screening of inbred animals . Most importantly, actual creation of mouse
models of diseases has been made possible by stem cell lines and methods
for introducing specific mutations into those cells , which has led to
an explosion of information . Unfortunately, mouse models are not ideal
for some human diseases. For example, in the mouse model of cystic fibrosis,
the mice fail to develop the lung and pancreatic pathology that are hallmarks
of the human disease, but have a more severe form of intestinal disease
. Furthermore, even though mice with improved disease features are being
developed through selective breeding , mice are still not ideal for many
purposes, especially those related to the evaluation of clinical interventions.
Thus, alternative animal models would be a boon for researchers. However,
in animals other than the mouse it has so far been extremely difficult
to develop embryonic stem cell lines that routinely give rise to viable
offspring .
In this paper we describe an alternative strategy for the discovery
of natural animal models of recessive genetic diseases. The strategy is
based on the hypothesis that disease frequencies across human populations
offer some guide to disease frequencies in animals. When disease
frequencies are high enough (>10-6), the method is feasible
using existing methods for genetic screening of genomic DNA.
The key to the feasibility of the method is the ability to screen for
unaffected heterozygotes. It is not sufficiently appreciated that even
rare, recessive genetic diseases have relatively high heterozygous gene
frequencies. For example, a recessive disease frequency of 1/1,000,000
arises from a carrier frequency of only 1/500. This enormous disparity
explains why carriers can be detected readily in populations for which
the associated recessive disease is apparently "non-existent".
In this chapter, we introduce the general concept, outline two attempts
to implement the approach , and provide a series of steps that should be
followed to allow this approach to become a general and cost-effective
alternative to stem cell technology.
1.2. Do Recessive
Human Genetic Diseases Have Animal Counterparts?
Some recessive diseases have been documented in both humans and animals
, but how likely is it that a specific human genetic disease will occur
in a specific animal species? The human genome is estimated to contain
between 30,000 and 40,000 genes . However, the Online Mendelian Inheritance
in Man lists fewer than 10,000 autosomal entries,
and fewer than half of these are recessive. The disparity between gene
number and disease number has many explanations, but the contribution of
each is unknown. If we consider only recessive mutations, we know from
experimental work that some of these cause early embryonic lethality when
homozygous while others cause no obvious phenotype. Another consideration
is that recessive diseases are usually so rare that the chance of a disease
escaping diagnosis is high. That leaves an unknown proportion of genes
for which it might be argued that the lack of a known disease state arises
simply because the mutation frequency in the associated gene is so low
that no human exists who has two copies of the mutated gene. How likely
is this?
The human population is estimated to be approaching 6 x 109
individuals. To estimate the number of mutations within this vast gene
pool we need to know the mutation rate for human genomic DNA. Unfortunately,
estimates of that rate vary widely. Based on extensive experiments with
Drosophila,
Crow gives an estimated mutation rate per nucleotide per generation, of
1.5 x 10-8, and predicts that the 3 x 109 nucleotide
pairs of the human genome, will therefore acquire ~100 new mutations in
each human zygote, with ~2% of these affecting genes. To avoid the accumulation
of an enormous mutational load, it is proposed that heterozygotes are mildly
but cumulatively disadvantaged, and that their preferential elimination
culls numerous mutations simultaneously to counterbalance the accumulation
. In contrast, experiments in which 50 independent lines of C elegans were
allowed to accumulate spontaneous mutations led to the conclusion that
the deleterious mutation rate per haploid genome was 0.0026 .
1.3. Carriers Greatly Exceed Affected Individuals.
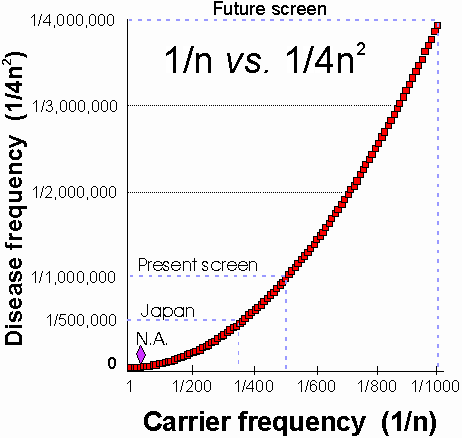
As stated above, the enormous disparity between heterozygote and homozygote
frequencies (Fig. 1) is not widely appreciated. Cystic fibrosis
(CF) illustrates some of the consequences of this disparity. CF has an
extraordinarily high frequency in the U.S. and northern European Caucasian
populations, where about 1/25 individuals are heterozygous for mutations
in the causative gene, CFTR. Cystic fibrosis is comparatively rare
in other populations. One estimate of the incidence rate of cystic fibrosis
in Japan gave a rate of 3.1 per million live births from 1969-1980 (~1/323,000).
The highest rates of CF were in Hokkaido (the most northern Island) and
lowest in Okinawa (the most southern island). The mean age at death from
CF was 3 years for both sexes during the period 1969-1985 . A similar estimate
of 1/350,000 using different methods was made more recently . Inspection
of Fig. 1 or simple calculation shows that the lowest estimate still corresponds
to a carrier frequency of ~1/295, suggesting that the Japanese population
(n » 108), has ~ 339,000 individuals
carrying disease-causing CFTR mutations. A similar exercise for
China suggests it has >4 million cystic fibrosis carriers.
The high incidence of cystic fibrosis in Caucasian populations results
primarily but not exclusively from the frequency of one very common allele
(D F508). In the CF population of the U.S. and
Canada, the D F508 mutation accounts for ~70%
of all alleles. Hence, if D F508 were to be
subtracted out, the frequency of cystic fibrosis in this population would
drop from about 1/2,500 (1/25 carrier frequency) to ~1/28,000 (~1/83 carrier
frequency). That is still much higher than the estimates for CF in Japan,
and suggests that factors other than the D F508
mutation are at work.
1.4. The Hypothesis: The Aggregate Background
Frequency Of Human and Animal Mutations are Similar.
We hypothesize that the aggregate frequency of non-D
F508 CF causing mutations in human populations offers a rough guide to
the aggregate CFTR mutation frequencies in non-human primates. This
hypothesis does not assume that any specific mutations found in human populations
will necessarily be found in non-human primates. Correspondingly, of course,
the frequency of particular mutations in the human population will not
provide information about particular mutation frequencies in non-human
primate populations; that is evident from the different pattern of mutations
observed in separated human populations. Thus, it does not make sense to
search non-human populations for specific mutations. Instead, a method
is required that is capable of detecting unknown mutations.
We know of no a priori reasoning and certainly no data that would
suggest a much different aggregate CF mutation frequency in non-human primates.
CFTR is a large gene and like any gene is susceptible to insertions or
deletions that cause frame shifts, as well as stop mutations and splicing
mutations. CFTR is also, for unknown reasons, extremely susceptible to
missense mutations that cause it to be misprocessed . Even wild type CFTR
is inefficiently processed. Approximately 75% of wild type CFTR protein
is degraded after core glycosylation and never reaches the plasma membrane--this
occurs across a range of cells expressing various levels of CFTR and so
is not merely an artifact of high levels of exogenous expression . At least
four critical regions in CFTR (the pore and the two NBFs) are susceptible
to missense mutations that interfere with CFTR's ability to function as
a Cl- channel¾ these also cause cystic
fibrosis . Finally, recent evidence indicates that some missense mutations
which have little affect on processing or chloride channel function can
cause CF by altering HCO3- transport.
In sum, the large size of CFTR creates many opportunities for mutations,
and a high proportion of all mutations render CFTR non-functional and lead
to disease in the homozygous state. In humans and mice, heterozygosity
for CF has no detectable disadvantage. Thus, leaving aside the possibility
of a heterozygote advantage, and barring some unforeseen feature that powerfully
selected against monkey carriers, there is no efficient mechanism to prevent
CFTR mutations from accumulating in a population at low frequencies, since
such frequencies will give rise to homozygotes too infrequently to alter
mutation frequencies in the population.
1.5 Which Species Should Be Studied?
The choice of species to be studied is greatly narrowed by several obvious
features. Because mice can be genetically manipulated, there is little
reason to study any species further removed from humans than mice. Among
remaining species, four main criteria determine suitability for discovery
of natural animal models. These criteria are (1) availability, (2)
experimental
tractability, (3) similarity to humans, and (4) genetic diversity.
The first two criteria need not be elaborated. However, the criterion of
human similarity can vary depending upon the disease of interest, such
that a more closely related species may be less optimal than species that
are further removed phylogentically. For example, sheep and pigs have lungs
that may be better experimental models of some human lung diseases than
monkeys. Genetic diversity within the target population is a crucial feature.
Unfortunately, the need for high genetic diversity excludes most domestic
populations of animals, but even wild populations may be unsuitable. For
example, cheetahs display an extreme degree of genetic homogeneity, presumably
as a result of a severe population bottleneck that occurred ~10,000 years
ago. .
Old world monkeys, particularly the genus Macaca, rank highly on all
4 criteria. (1) Availability is good. Wild populations are still large
(though declining at an alarming rate) and are extensively distributed
throughout Africa and Asia. An estimated 40,000 primates are imported annually
into the U.S for research purposes. Of greater relevance are the large
number of primates (~16,000) maintained at NIH Regional Primate Research
Centers. This population is bred exclusively for research, and the monkeys
receive excellent care and typically live for longer than a decade. The
last point is critical, because it is essential to be able to retrieve
an animal after a mutation has been identified in its DNA. (2) Monkeys
are good experimental subjects, and for some experiments are virtually
the only suitable animal subjects. (3) Monkeys are in general more similar
to humans than any other species except the great apes, and apes have become
so endangered that their use is virtually precluded except for the most
essential studies. (4) Finally, with regard to genetic diversity, the evidence
suggests that monkeys may be an unusually rich repository of genetic diversity.
Studies of 6 species of macaques and of 23 local populations of Rhesus
monkeys spread across Vietnam, Burma, and 10 provinces of China extended
previous estimates of genetic heterogeneity among and within species. Our
own studies have confirmed a high degree of genetic diversity even within
Macaca
maintained for many years within Regional Primate Research Centers.
A possible heterozygote advantage. With regard to a possible
heterozygote advantage, it may be relevant that monkeys are notoriously
susceptible to secretory diarrhea. Human CF heterozygotes are thought to
be partially protected against certain diarrheal diseases because CFTR
is rate-limiting for Cl--mediated electrolyte and fluid secretion from
intestinal crypts . In some secretory pathways, fluid secretion by CF heterozygotes
is indeed reduced to 50% of normal . Hence, diarrheal diseases that stimulate
the CFTR-dependent pathway should cause less fluid and electrolyte loss
in heterozygotes. This hypothesis has been tested directly by administering
cholera toxin to heterozygous CF mice, but results were conflicting .
A study of serum electrolyte values in 100 rhesus monkeys with diarrhea
observed hyponatremia in 88% and hypochloremia in 80% . This strongly suggests
that the putative protective effect of CFTR mutations should also apply
to non-human primates, and could result in positive selection pressure
and hence some enrichment of CF alleles in non-human primate populations.
To give some indication of the magnitude of this potential selection pressure
for CFTR mutations, in the California Primate Research Center, 34% of non
experimental deaths in macaques one year of age and older were due to gastrointestinal
disease .
Possible heterozygote disadvantages. The severity of disease
caused by CFTR mutations is closely related to the extent to which
CFTR-mediated Cl- conductance is lost. Mild mutations can arise
for each class of CFTR mutation; for example, some trafficking mutations
allow a certain proportion of CFTR to be processed , some regulatory mutations
do not completely disrupt function , and all conductance mutations to date
produced only a partial loss of conductance.
Within a critical range of residual CFTR function, subjects no longer
display the classic cystic fibrosis syndrome, but instead suffer, if they
are male, from sterility secondary to congenital bilateral absence of the
vas deferens (CBAVD) . The extreme susceptibility of the vas deferens to
mutations in CFTR is not completely understood. However, part of answer
may be that CFTR is spliced differently in the vas. A common mutation
that contributes to CBAVD is a reduction in a tract of 8 thymidines within
intron 8 to a 5-thymidine variant that leads to missplicing of CFTR. The
proportion of misspliced CFTR is greater in the vas deferens than in the
lung . Unlike humans, mice homozygous for CFTR mutations remain fertile
. Given this species difference, a possibility that must be considered
is that in some species male fertility will be lost or compromised even
in the heterozygous state.
````````
1.6. Testing the Approach: the Search for a
Monkey CF Carrier.
In spite of such arguments, only direct experimental test can provide
accurate estimates of the frequency of CFTR mutations and polymorphisms
in a given species. With no additional information, the chances of mutations
being more frequent in non-human primates than in human populations is
equal to the chance that they are less frequent. For a mutation frequency
of 1/500 (for a disease frequency of 1/1,000,000); screening 1,500 animals,
yields ~95% chance of detecting a mutation if the detection method is perfect.
Of course no detection method is perfect. The SSCP method is near perfect
for detecting small insertions and deletions, and probably detects >90%
of point mutations. However, it does not detect intronic mutations or deletions
of entire exons. Based on assays of CF populations, the SSCP/HD method
we use is estimated to be able to detect ~95% of CF mutations . Thus, screening
of 1,500 primates provides a 95% chance of finding a mutation if mutations
occur at a frequency of 1/400 or greater, equal to a disease frequency
of ~1/640,000. It is worth emphasizing that even a disease frequency of
1/100,000 would make it unlikely that even a single CF birth would have
occurred among the entire primate population in all of the U.S. Primate
Research Centers during the last decade. Given the infant mortality rate
mentioned above, even if such a rare event occurred, the chance that it
would have been detected is remote. This emphasizes the power of heterozygote
analysis even among populations in which the disease appears to be "non-existent".
The general significance of this program will be to determine
the feasibility of establishing animal models for any disease by
screening. If our hypothesis of an approximate correspondence in mutation
frequencies among primates is confirmed, a program like the one we propose
should be at least as cost-effective as the production of mice by stem-cell
recombinant methodology.
-
Materials
2.1. Whole blood (~ 3 ml) was
obtained by venipuncture mainly during routine medical checkups of primates
and was shipped on ice in EDTA-containing (purple top) tubes.
2.2. EDTA - Ethylenediaminetetraacetic
acid ( Sigma, St. Louis, USA) 1 ml of 10% EDTA solution was used for each
blood sample.
2.3. Puregene DNA isolation
kit (Gentra systems, Inc., Research Triangle Park, North Carolina, USA).
2.4. CFTR Primers, Operon technologies,
Inc (For primer sequences, see Wine, 1998 ).
2.5. AmpliTaq® DNA Polymerase
and GeneAmp® 10X PCR Buffer was used for PCR amplification (Applied
Biosystems, Foster city, CA, USA).
The GeneAmp 10X PCR Buffer is composed of 500 mM potassium chloride,
100 mM Tris-HCl (pH 8.3 at room temperature), 15 mM magnesium chloride
and 0.01% (w/v) gelatin GeneAmp® dNTPs GeneAmp® dNTPs deoxynucleoside
triphosphates were used Composition: Each of the 4-vial set contains 320
µL of a 10 mM Solution of either dATP, dCTP, dGTP or dTTP.
Isotope a 32P dCTP (3000 Ci/mM) (Amersham
Pharmacia Biotech, Piscataway, NJ, USA) 0.5 mCi of a
32P dCTP (3000 Ci/mM) isotope was included in each 10 m
L PCR mixture for labeling.
2.6. Denaturing mixture : 95%
formamide, 20 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol, and
20 mM NaOH
Formamide > 99.5 GC ( Sigma, St. Louis, USA)
EDTA - Ethylenediaminetetraacetic acid ( Sigma, St. Louis, USA)
Bromophenol blue ( 3,3,5,5-tetrabromophenolsulfonephthalein sodium
salt)
(Sigma, St. Louis, USA)
Xylene Cyanole FF (dye content: ~75%) (Sigma, St. Louis, USA)
NaOH Sodium hydroxide ( Sigma, St. Louis, USA)
2.7. Standard components for
Vertical polyacrylamide Gel Electrophoresis (e.g. MDE gel solution or acrylamide/bis-acrylamide
mix, glycerol, TEMED, vertical slab gel KODAK BIOMAX STS 45i apparatus
for running 35 x 40 cm, 0.4 mm-thick gels).
Plates (35x40cm ) for vertical slab gel KODAK BIOMAX STS 45I apparatus
(Eastman Kodak Company, Rochester, New York, USA).
TEMED (N,N,N,N-tetramethylethylenediamine ) >99% ( Sigma, St. Louis,
USA).
MDE gel (BioWhittaker Molecular Applications, Rockland, ME , USA)
Glycerol >99% Electrophoresis reagent ( Sigma, St. Louis, USA) 10% added
to MDE gel solution.
Polyacrylamide: Bio-Rad Laboratories 40% acrylamide : N,N-Methylenebisacrylamide
solution, 37.5:1 (2.6%C).
TBE buffer (TRIS-BORATE-EDTA Buffer) 5X concentration (Sigma, St. Louis,
USA).
1 liter of 1 X TBE buffer was used for electrophoresis.
Electrophoresis power supply EPS 1001 (Amersham Pharmacia Biotech, Piscataway,
NJ, USA) was used for electrophoresis.
Autoradiography was done on KODAK Scientific Imaging Film X-OMAT AR
(35x43 cm) (Eastman Kodak Company, Rochester, New York, USA).
2.8 Mutations were made with
Stratagene's Quick-change site- directed mutagenesis
kit. (La Jolla, CA) and verified with restriction
enzymes (Life Technologies, Grand Island, NY) or sequencing.
2.9. Expression:
Plasmid purification: Qiagen plasmid maxi kit (Valencia, CA)
Transfection: SuperFect transfection reagent. (Qiagen, Valencia, CA)
Dish coating: fibronectin (Sigma, F2006) (St. Louis, MO)
DME H21, 10% fetal bovine serum, 2 mM glutamine, and Pen/Strep (100
U/m g/ml). (Sigma, St. Louis, MO)
2.10 Functional assay. Efflux
buffer was (in mM): 50 N-2-hydroxy ethylpiperazine-N'-2-ethane sulfonic
acid (HEPES), 5.4 KCl, 130 NaCl, 1.8 CaCl2, 1.0 Sodium
Phosphate (monobasic), 0.8 MgSO4, pH adjusted to 7.4
with NaOH, and glucose 100 mg/100 ml, all from Sigma, and 125I
/ml (St. Louis, MO).
-
Methods.
The overall approach is outlined in Supported by the Cystic Fibrosis Foundation, by NIH HL51776, and by
RR00169 to the California Regional Primate Research Center. We thank the
staffs of the Primate Research Centers in California, Louisiana, Oregon
and Washington; especially Jenny Short, Phil Allen, Ron Walgenbach, Margaret
Clarke, Mark Murchison, Steve Kelley and Debra Glanister. S. Vuillaumier,
INSERM, Paris supplied the sequence of exon 1 and flanking segments from
several primate species. We thank Ron Kopito and Cristi Ward for supplying
293 cells, the pRBG4 vector, CFTR-pRBG4, and help with transfection protocols.
Numerous individuals assisted with SSCP and functional analysis, especially
Gregory Hurlock, Eugene Kuo, Mauri Krouse, Clare Robinson, Margaret Lee,
Uros Potocnik, and Metka Ravnik-Glava¹
.